Single Cell Multiomics
Overview
Single cell multiomics is a next-generation sequencing (NGS) application that performs RNA-seq and ATAC-seq on the same cell simultaneously to reveal insights into the epigenetic regulation of gene expression.
General Workflow
A typical single cell multiomics experimental workflow involves the isolation of single intact cells. After the cells are isolated, they are permeabilized to allow entry of Tn5 transposase enzyme, which tagments open chromatin regions. The cells are then lysed and mRNA is captured with oligo dT magnetic beads. After magnetic separation, the tagmented genomic DNA is purified, and PCR extension and amplification is performed to create an NGS library. The mRNA captured on oligo dT beads is reverse transcribed and the subsequent mRNA/cDNA hybrids are directly tagmented by Tn5. After initial end extension, the tagmented cDNA is amplified with indexed PCR to prepare the mRNA-seq library.
Data Analysis
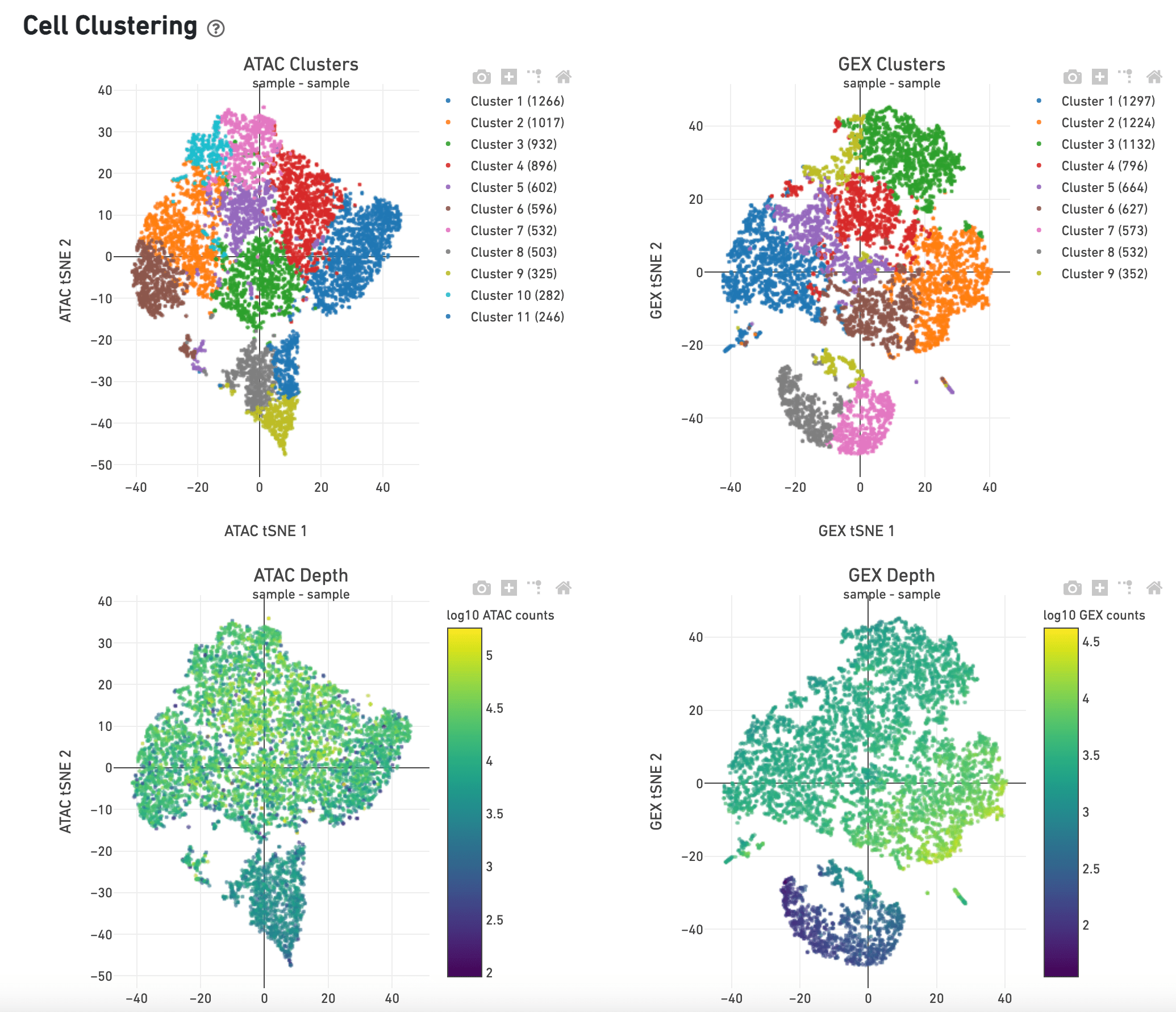
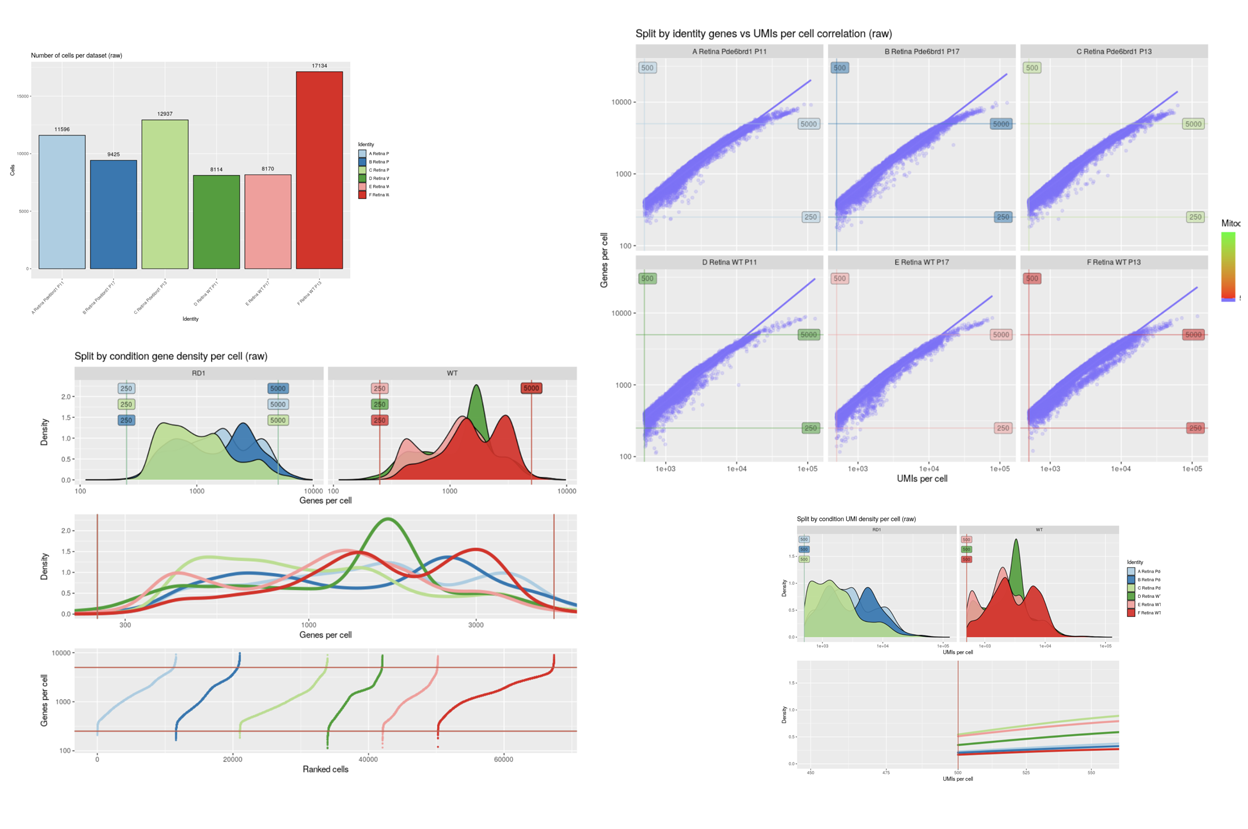
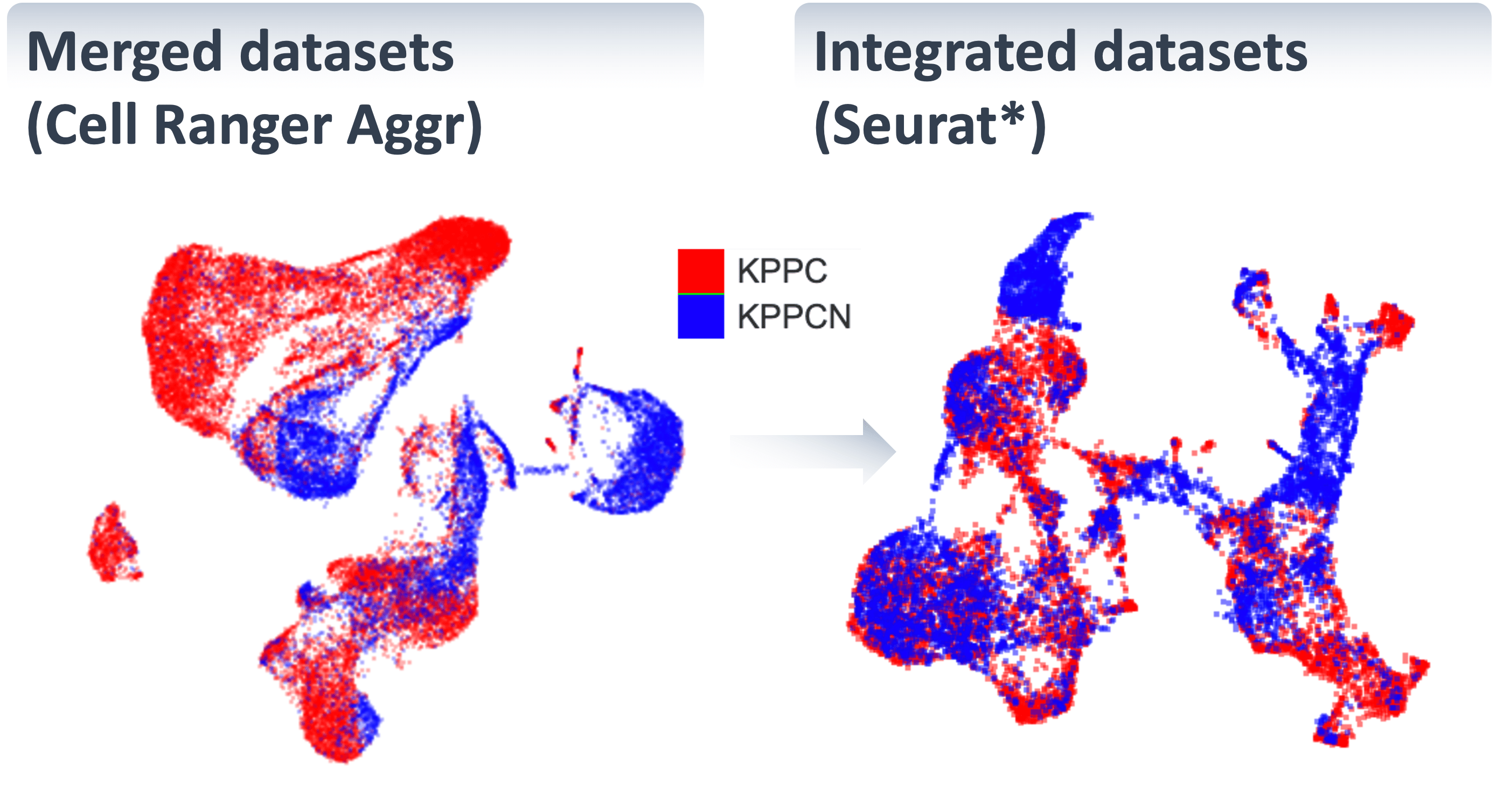
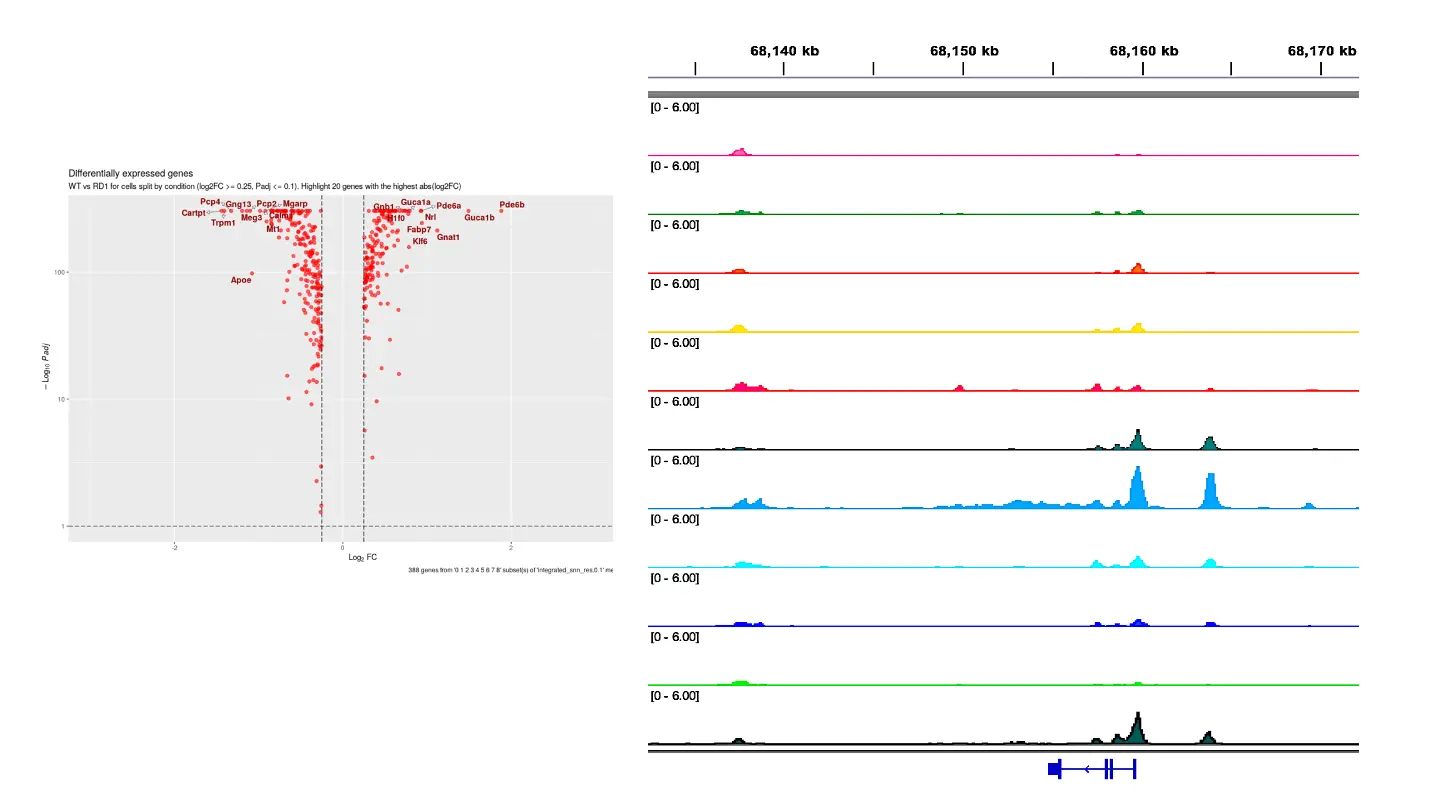
SciDAP is a no-code bioinformatics platform that enables scientists to analyze NGS-based data without a bioinformatician. It has a collection of 18 built-in pipelines based on open-source workflows to analyze data from single cell multiomic libraries. The set includes pipelines for initial processing of scRNA-Seq and scATAC-Seq data, aggregation of multiple samples and filtering out bad cells, clustering and differential expression, and abundance estimation.
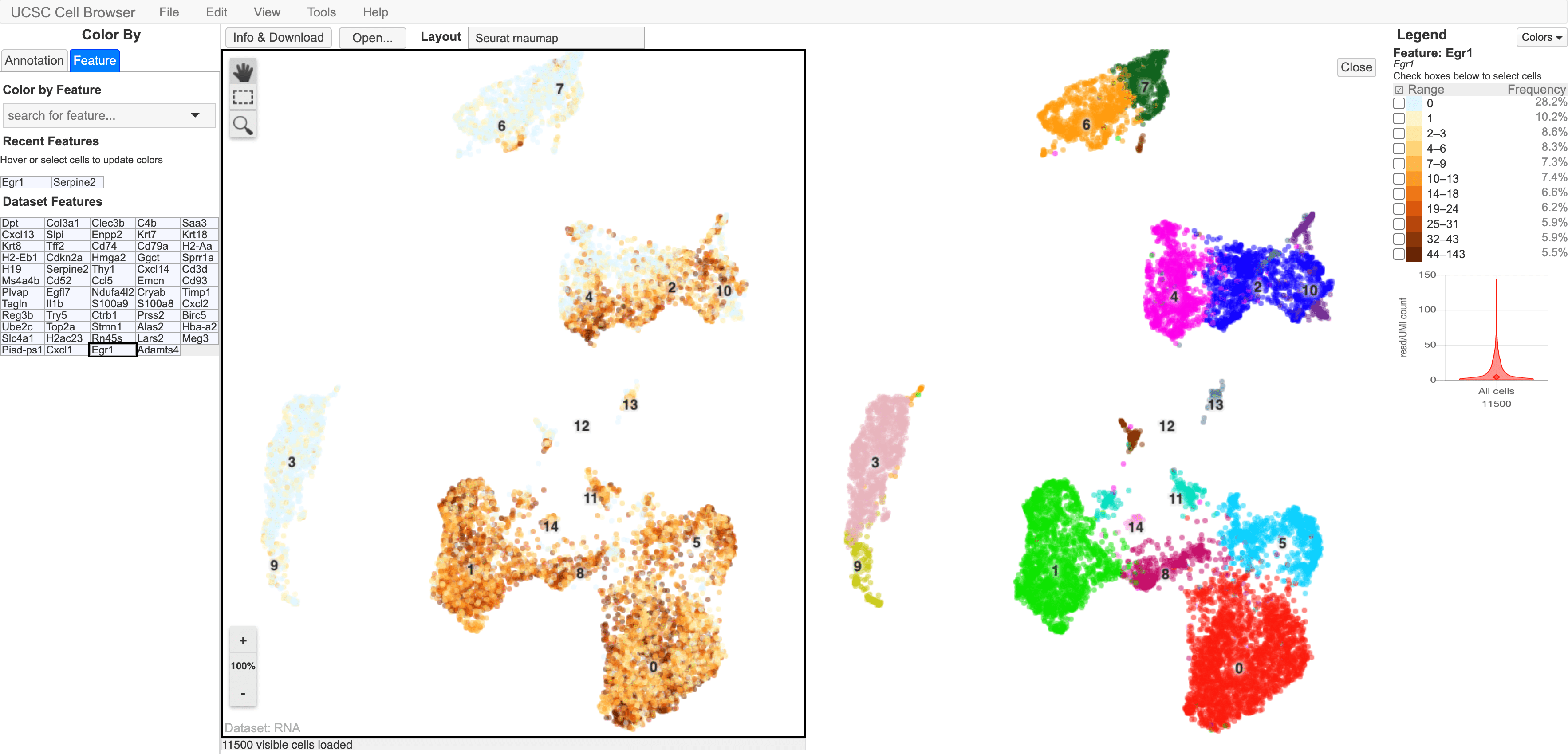
SciDAP starts from the FASTQ files provided by most DNA core facilities and commercial service providers. Starting from raw data allows SciDAP to ensure that all experiments have been processed in the same way and simplifies the deposition of data to GEO upon publication. The data can be uploaded from the end-users’ computer, downloaded directly from an FTP server of the core facility by providing a URL, or from GEO by providing SRA accession number. The processing results can be visualized on the UCSC cell browser.